《现代物理有机化学》笔记:有机电子材料
17.1.1 无限体系:特别的聚乙炔
首先绘制出“聚乙炔”的能级图:
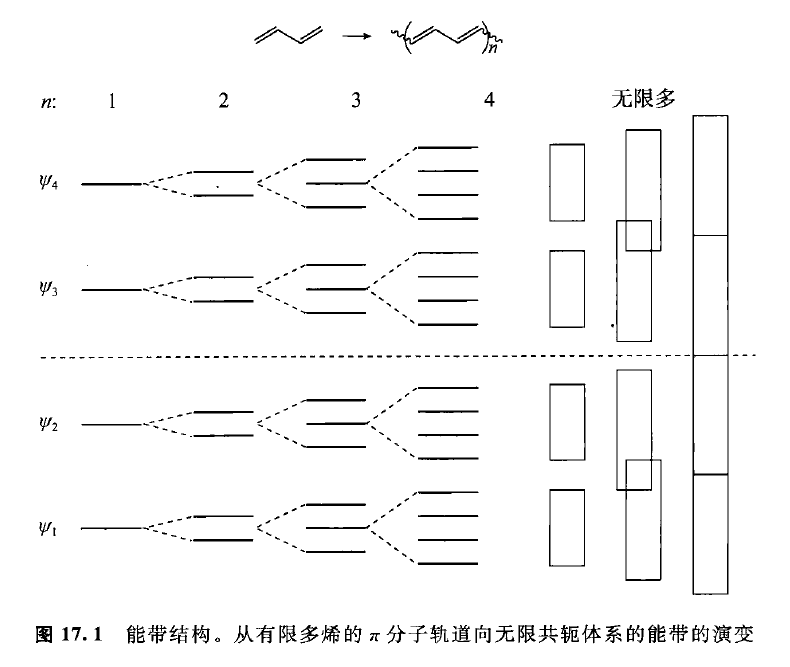
而我们处理的策略就是将线性体系以环多烯的方式处理,因为它们的电子结构差不多,它们的能级满足以下关系:,其中为轨道节点数目,为原子总数目。例如,我们分析环戊二烯自由基():
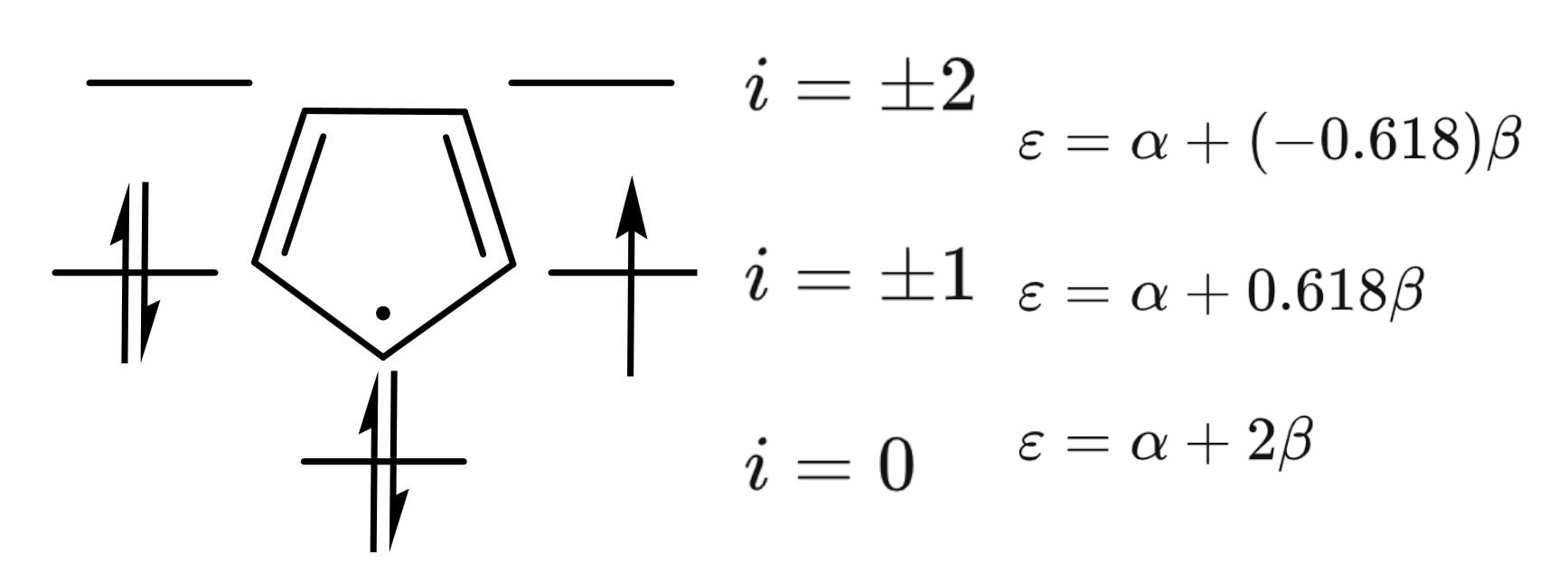
其中,时的轨道是反键轨道,其余的是成键轨道。那么,当$N\to + \infty \epsiloni$变化的图像:
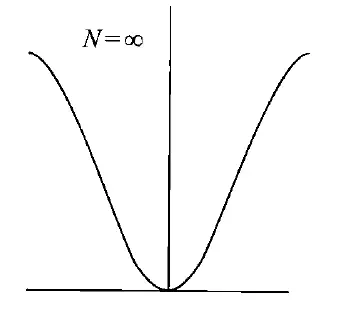
可以看出,它类似于一个正弦曲线,最小值为,最大值为。
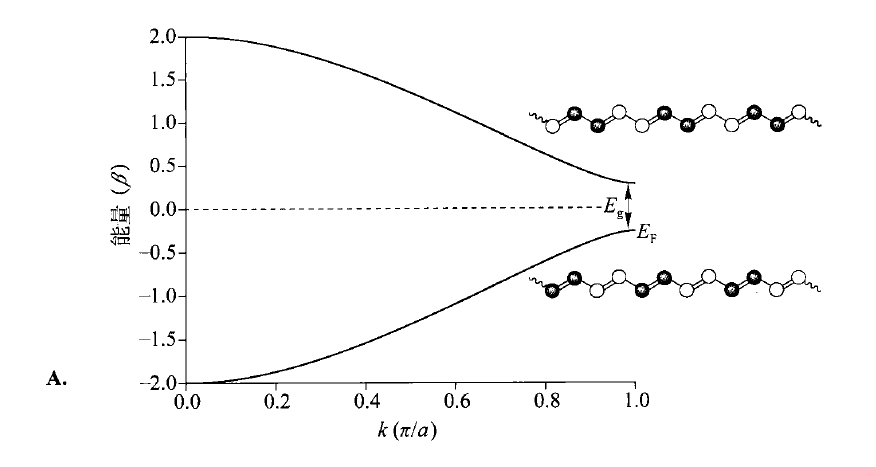
倘若我们将固体化学的概念引入这里,比如“晶胞”,即“重复单元”,然而,无限体系中的所有$\mathrm{CH} \mathrm{CH} a\mathrm{CH} kk=\frac{2\pi i}{Na} -\frac{N}{a} \le i\le +\frac{N}{a}-\frac{\pi}{a} \le k\le +\frac{\pi}{a}k\epsilon\epsilon=\alpha+\cos( ka )\times\beta$,则可以作出相应的图像:
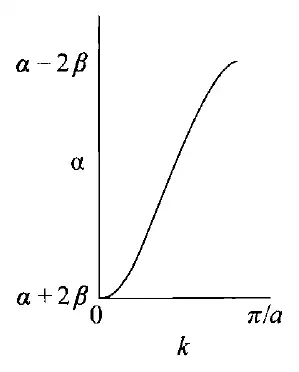
代表着能带内“相”的关系,代表着“相空间”,即轨道相位组合的方式。时,所有单体的相位相同,$k=\frac{\pi}{a} $时,所有单体的相位相反。
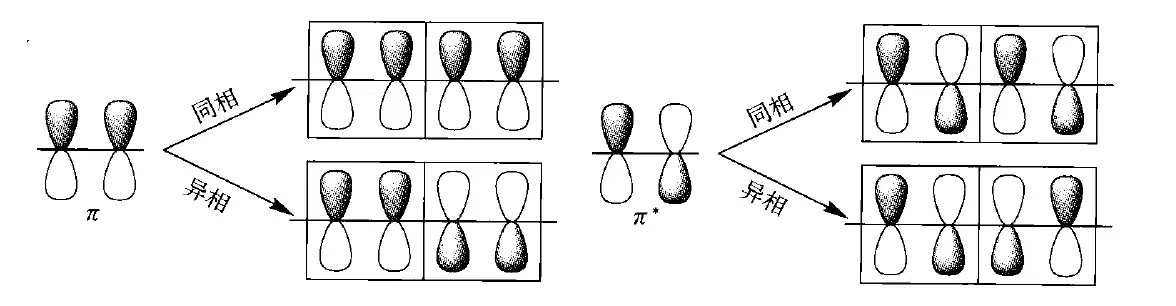
时,所有相位相同,此时能量应最低,为; 时,所有单体的相位相反,能量最高,为;而π轨道(如上图中左下侧)异相组合时,能量应呈现非键轨道特性,为。(注意:这里原书认为单体,即重复单元为两个$\mathrm{CH} $,那么相应地,单体长度也会扩大两倍,变成 ,)
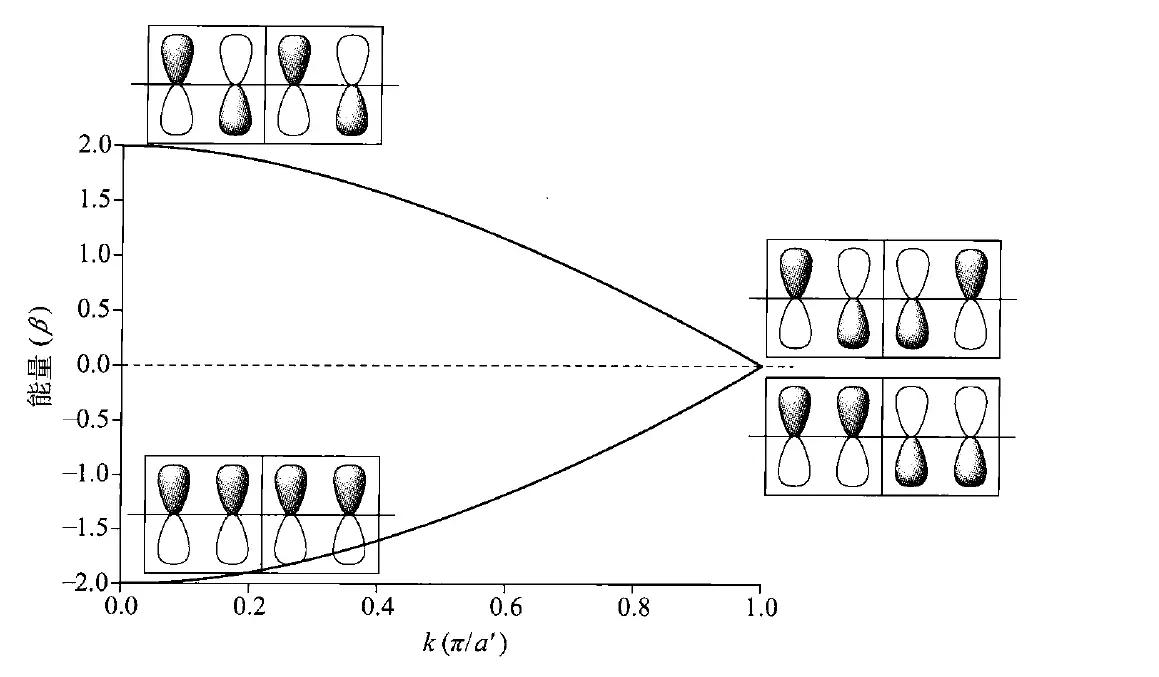
不过,不管怎么取“晶胞”,分子的电子结构都不会改变,那么理论上,聚乙炔的π轨道应当全满, 轨道应当全空,其Fermi能级(,和HOMO是一个意思)就在中间,能量为 。但在这个模型中,能带( 和 轨道组成的)之间的能级差(,能隙)应该为 ,所以,聚乙炔就应该是一种有机金属导体了!可惜事实真的如此吗?
更何况,真实的材料可不会是一维的,所以以上分析还是有点瑕疵。这也是Hückel理论的一个缺陷。
17.1.2 Peierls形变
我们发现,在Hückel理论中,聚乙炔的能隙等于,而环丁二烯也是一个类似的体系:
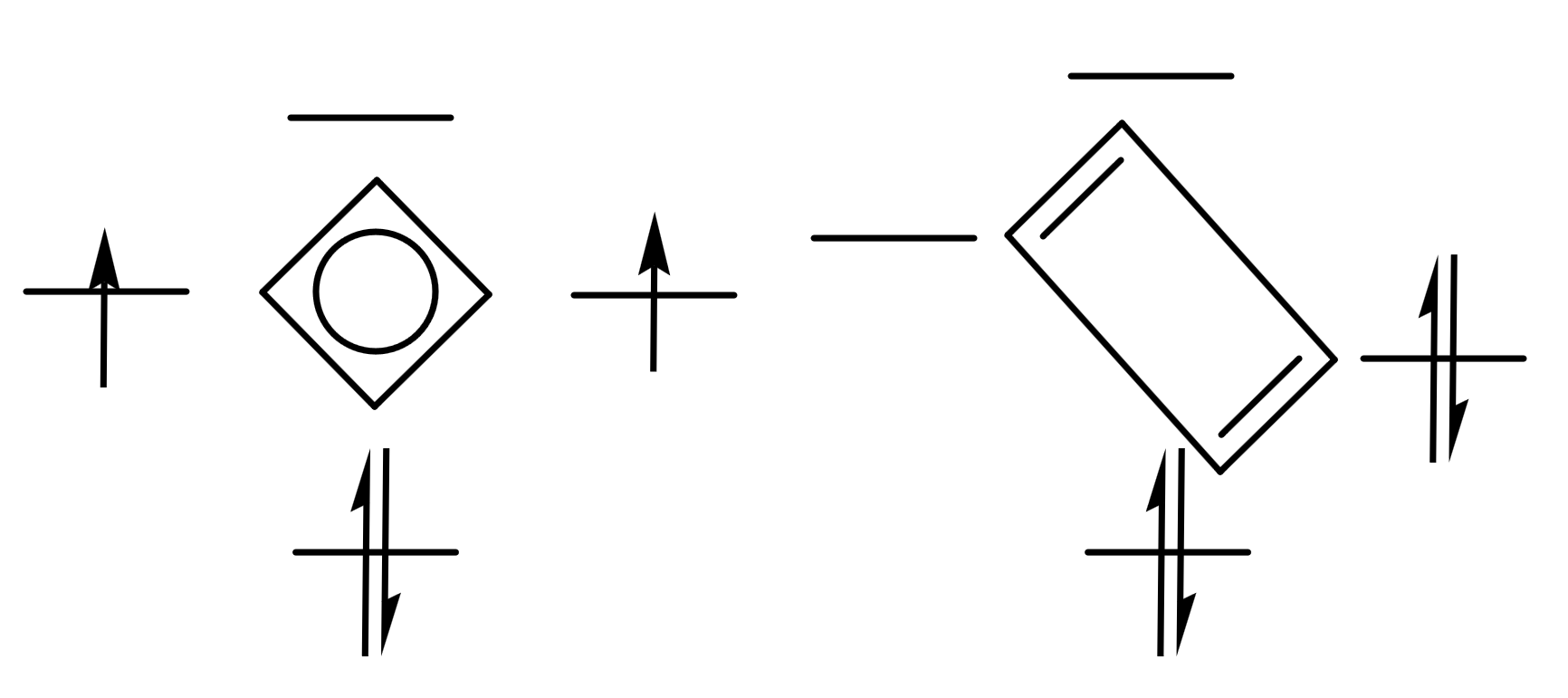
如果不谈反芳香性等等因素的话,那环丁二烯本来应该是正方形的,这没什么问题。但是它实际上处于一个矩形的构型(长键长,短键长),这就导致HOMO和LUMO裂分,从而变得更稳定——矩形的构型能量比正方形低——这也可以被称作是“伪Jahn-Teller效应”。因此,环丁二烯的基态应该是单重态(上图右),而非三重态(上图左),正方形的构型其实算是两种矩形构型交替变化的一个过渡态罢了。
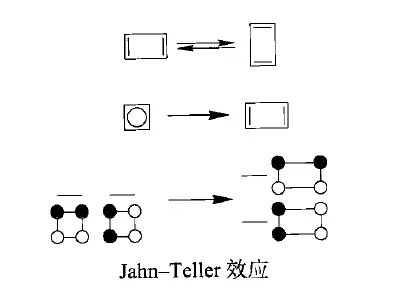
类似的效应在聚乙炔中也出现了,按照我们的预期,聚乙炔的C-C键应该都是等长的——可事实却不是这样,它的键长发生了形变,变成了长短交替的结构。几何形变让它出现了能隙,,所以实际上,聚乙炔是一种半导体,这种形变叫做Peierls形变。
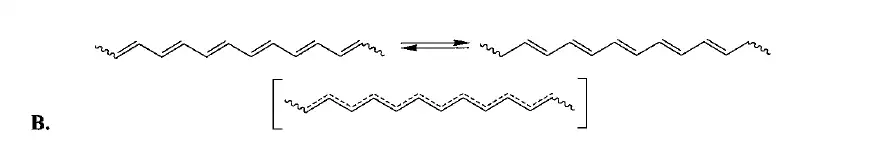
此时,聚乙炔的HOMO(也就是)是低于的,确实是稳定化了。其实在多数中性、闭壳的体系中,这种长短键交替的现象是比较常见的。苯算是一个特例,由于强键的束缚,它的键长的确是均一的。
17.1.3 掺杂
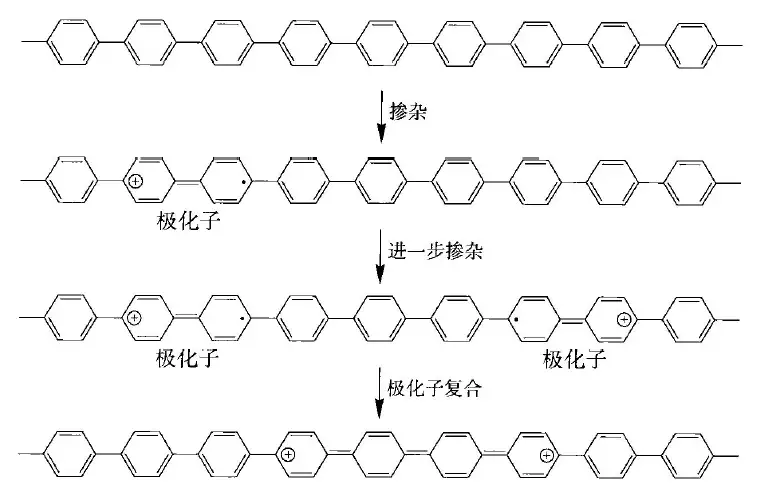
在体系共轭聚合物中,聚乙炔等等只能算是半导体,而绝缘体更是普遍的情况。所以,为了改善它们的导电性,掺杂是一个很好的办法。例如把聚合物置于氧化剂(如,)氛围中,让其失去电子,引入正电荷,进行p型掺杂;或是用还原剂(如金属钠,萘-钠)处理进行n型掺杂。那么我们先讨论p型掺杂。
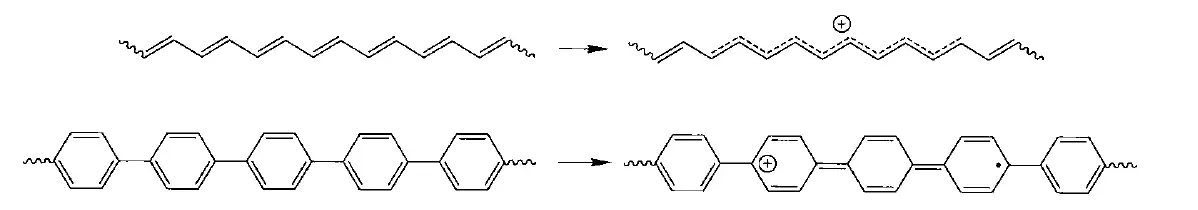
进行n型掺杂,引入正电荷或自由基后会产生一个部分离域的正离子,为什么是部分离域呢?
我们知道,烯丙基自由基比乙基自由基稳定,是因为它有离域体系,然而随着离域体系越来越大,自由基应该是更稳定的,随着碳原子数的增加,例如戊二烯基自由基,肯定是比烯丙基自由基稳定的,但是它们之间能量的差值会越来越小,到最后,离域提供的稳定化能只能说是微乎其微,不足挂齿了。(可能可以用共轭饱和理论来解释)
另外,由于Peierls形变为聚合物提供了稳定化能,但是掺和进来的正离子或自由基打乱了这个节奏,离域的能量不足以补偿打乱Peierls形变抬升的能量。
随着掺杂得越来越多,产生的部分离域的电荷和自由基(称作“极化子”)也会变得越来越多,使得电导率上升。
这样掺杂的一个结果,就是能够形成“中间能隙束缚态”,代表着电子的缺失,也可以看作空轨道的引入,单占轨道从价带中独立出去。它的另一种解释是极化子的形成消除了Peierls形变。并且,新轨道的产生使得光学跃迁成为了可能,让产生的聚合物带上了颜色。
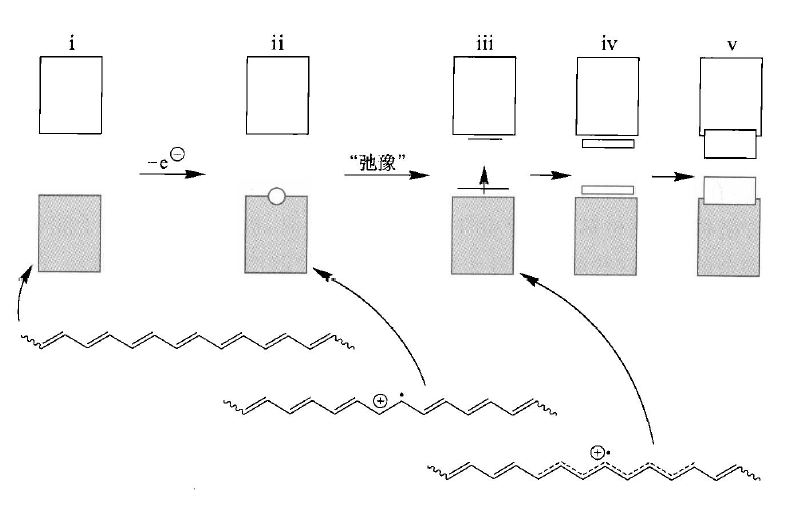
重度掺杂也会导致“双极化子”的形成,它可能是两个极化子复合而成的。如果极化子是自由基,则它们会复合成新键;若是电荷,库仑斥力不应该会阻碍它们的复合吗?在气相中的确是这样的,但在固相中,材料的介电系数更大,减小了库仑斥力,并且引入的阴离子,如 、等,扩散能力较强,屏蔽了其电荷,促使双极化子产生。
与金属导体不同,这些聚合物的电导率(,电阻率的倒数)常常与温度反相关,这是因为极化子的移动会导致碳骨架(它们的键长、键角等)轻微地改变,这需要一定的热活化能。金属在低温时,干扰晶格中自由电子运动的振动会减少。综上,电导率与温度的关系也是判定是否为金属导体的一大标准。
以上是对于p型掺杂半导体的论述,同理,n型掺杂也是差不多的。
致谢
特别感谢@lesscho 对本文的斧正,感恩捏!
原文发表在知乎。